1 Servicio de Histopatología.2 Servicio de Neumonología.Hospital Británico. Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2017;47(4):259-268
Recibido: 30/11/2016 / Aprobado: 15/05/2017 / Publicado en www.actagastro.org el 18/12/2017
Resumen
El carcinoma hepatocelular (CHC) es un tumor con diferenciación hepatocítica que normalmente no expresa marcadores de origen biliar como las citoqueratinas (CK) 7 y 19. Sin embargo, existe un subgrupo de CHC con expresión de marcadores de ambos linajes, que se desarrollarían a partir de células hepáticas progenitoras (CHP) y tendrían peor pronóstico. La prevalencia de la expresión de CK y de otras proteínas relacionadas a la hepatocarcinogénesis no ha sido explorada en series latinoamericanas. Objetivo. Caracterizar una serie argentina de CHC, y analizar la expresión de CK, E-cadherina, ß-catenina y p53. Métodos. Se analizaron retrospectivamente 126 piezas quirúrgicas, 51 de ellas con CHC. Resultados. Se encontró mayor prevalencia de CHC en el género masculino (75%), con edad media de 63 años y mayormente asociados al virus de la hepatitis C (39%). Las técnicas de inmunohistoquímica revelaron positividad con CK8/18 (86%), Hepar-1 (69%), Glypican-3 (66,6%), CK7 (52,9%), CEA (22%), CK19 (17,6%), E-cadherina (89%), ß-catenina (20%), y p53 (24%). Conclusión. En nuestra casuística, un alto porcentaje de CHC mostró positividad con CK7 y CK19. La tendencia al mayor índice proliferativo en los CHC positivos para dichos marcadores, así como el menor tamaño y la ausencia de invasión vascular en los tumores negativos para CK7, señalarían un comportamiento más agresivo en los CHC con expresión de marcadores biliares, potencialmente derivados de CHP. Estudios más amplios serían necesarios con el fin de dilucidar estos interrogantes y su relación con los diferentes agentes etiológicos involucrados.
Palabras claves. Carcinoma hepatocelular, hepatocarcinogénesis, célula progenitora hepática, citoqueratina.
Morphological and immunohistochemically characterization of hepatocellular carcinoma in Argentina. Relevance of the expression of cytokeratin and other proteins related to hepatocarcinogenesis
Summary
Hepatocellular carcinoma (HCC) is a tumor characterized by hepatocyte differentiation that usually does not express biliary markers, such as cytokeratin (CK) 7 and 19. However, there is a subgroup of HCC that express markers of both lineages and that may arise from hepatic progenitor cells (HPC) and have a worse prognosis. The prevalence of expression of CK and other proteins related to hepatocarcinogenesis has not been evaluated in Latin-American series. Aim. The aim of this study was to characterize an Argentinean series of HCC and analyze CK, E-cadherin, ß-catenin, and p53 expression. Methods. 126 surgical specimens, of which 51 had HCC, were analyzed. Results. A higher prevalence of HCC was found in male patients (75%), with mean age of 63 years old. Hepatitis C virus infection was the most common etiology (39%). Immunohistochemistry techniques showed positivity for CK8/18 (86%), Hepar-1 (69%), Glypican-3 (66.6%), CK7 (52.9%), CK19 (17.6%), CEA (22%), E-cadherin (89%), ß-catenin (20%), and p53 (24%). Conclusion. In our series, a high percentage of HCC was positive for CK7 and CK19. A trend towards a higher proliferative index in HCC that was positive for these markers as well as smaller size and absence of vascular invasion in CK7-negative tumors, point to a more aggressive behavior of HCC with biliary-marker expression that may derive from HPC. Larger studies would be necessary to elucidate these issues and their association with different etiological agents.
Key words. Hepatocarcinoma, hepatocarcinogenesis, hepatic progenitor cell, cytokeratin.
AbreviaturasCHC: carcinoma hepatocelular.VHB: virus de la hepatitis B.VHC: virus de la hepatitis C.HH: hemocromatosis hereditaria.Hepar-1: hepatocyte paraffin 1.CK: citoqueratina.CHP: células hepáticas progenitoras.OMS: organización mundial de la salud.CEA: antígeno carcinoembrionario.CRIHB: Comité de Revisión Institucional del Hospital Británico de Buenos Aires.
El carcinoma hepatocelular (CHC) representa el tipo histológico más común entre los tumores hepáticos primarios y la tercera causa de muerte por cáncer en el mundo.1-3 Recientemente, se ha descripto un incremento de su incidencia y agresividad, que confieren a los pacientes un pronóstico desfavorable, por lo que la detección precoz constituye un verdadero desafío. La infección crónica con el virus de la hepatitis B (VHB) o C (VHC) y el abuso de alcohol se reconocen como las causas más frecuentes de CHC en un contexto de inflamación crónica y cirrosis.2-4 No obstante, otras condiciones como la esteatohepatitis no alcohólica, la hemocromatosis hereditaria (HH) y las hepatopatías autoinmunes, entre otras, han sido reportadas en asociación con dicha neoplasia.1-4
En Argentina, ha sido previamente analizado el impacto epidemiológico de los CHC.5-7 Sin embargo, no se han reportado estudios morfológicos que describan el histotipo y el inmunoperfil de esta neoplasia. A nivel histológico, se reconocen diferentes patrones arquitecturales y variantes citológicas que han dado lugar a distintas clasificaciones, destacando el alto grado de polimorfismo de estas lesiones.1, 8, 9 Esta amplia variedad inter e intratumoral comprende un espectro de CHC que abarca desde tumores bien diferenciados hasta aquellos con pobre diferenciación, por lo que la estirpe celular resulta difícil de establecer. En este contexto, los hallazgos obtenidos por inmunohistoquímica aparecen como herramientas complementarias de utilidad diagnóstica.9
En el hígado normal, los hepatocitos expresan Hepar-1 (Hepatocyte paraffin 1) y CK 8/18 (citoqueratina 8/18), mientras que el epitelio biliar expresa CK7 (citoqueratina 7) y CK19 (citoqueratina 19). Sin embargo, también existen células hepáticas progenitoras (CHP) o stem cells, que coexpresan marcadores hepatocíticos y biliares, que tendrían el potencial de diferenciarse hacia cualquiera de los dos linajes. La marcación positiva con CK7 y CK19 en algunos CHC sugeriría un posible origen en las CHP, a partir de las cuales se desarrollaría un subgrupo de CHC con peor pronóstico.10
Las técnicas de inmunohistoquímica cobran un interés suplementario en el estudio de moléculas relacionadas a la carcinogénesis y/o al crecimiento tumoral, cuya expresión en CHC resulta controversial en la actualidad.11-13 Un ejemplo claro lo constituye la disminución en la expresión de las moléculas E-cadherina y ß-catenina, así como la mutación del gen supresor de tumores p53, clásicamente descriptas en tumores epiteliales de distinta estirpe.14-20 De esta manera, el desafío de las clasificaciones futuras radicaría en el aporte de datos histopatológicos e inmunofenotípicos que contribuyan a predecir el comportamiento indolente o agresivo de dichas neoplasias.21
El objetivo de este trabajo fue caracterizar una serie argentina de CHC a nivel morfológico e inmunohistoquímico, con especial énfasis en la expresión de CK7 y CK19 en relación con el linaje celular, y analizar la expresión de otras proteínas implicadas en la hepatocarcinogénesis.
Material y métodos
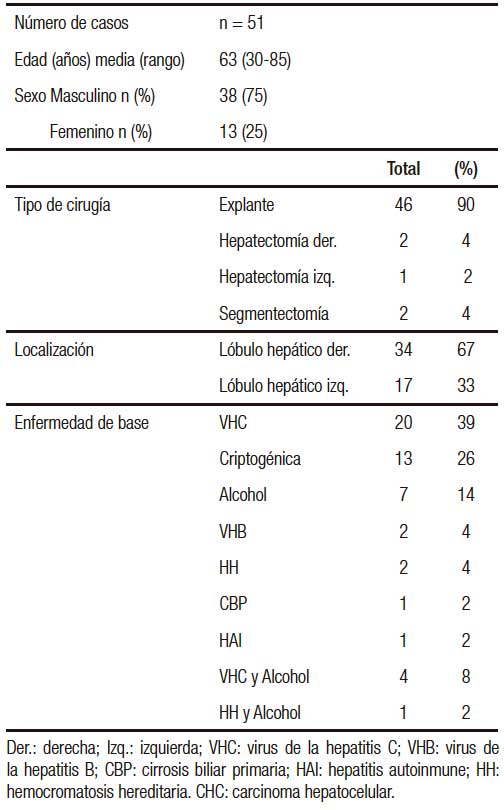
DiseñoSe analizaron en forma retrospectiva 126 piezas quirúrgicas hepáticas de pacientes operados entre agosto de 2010 y mayo de 2014. Se seleccionaron los especímenes con CHC (n = 51): 46 hepatectomías totales (explantes) y 5 resecciones parciales. Se registró la edad y el género de los pacientes. El presente estudio fue aprobado por el comité de revisión institucional del Hospital Británico de Buenos Aires (CRIHB) de acuerdo con los principios de la declaración de Helsinki.
CriteriosSe incluyeron los casos de CHC con etiología asociada conocida (VHC, VHB, etilismo crónico, cirrosis biliar primaria, hepatitis autoinmune, HH) y los de causa indeterminada (criptogénica). Se excluyeron los explantes hepáticos de causa no tumoral, los carcinomas hepatocelulares fibrolamelares y los colangiocarcinomas.
MuestrasEn las piezas quirúrgicas se analizó la localización, tamaño y multicentricidad tumoral. Se agrupó a los tumores iguales o mayores de 3 cm como CHC de gran tamaño y a los menores de 3 cm como CHC pequeños.
Procesamiento de las muestrasA partir del material fijado e incluido en parafina se realizaron cortes histológicos y se colorearon con hematoxilina y eosina. Todos los especímenes fueron obtenidos del archivo de patología del Hospital Británico de Buenos Aires. El grado histológico fue evaluado según la clasificación de Edmondson y Steiner (grados I a IV) y según su diferenciación, de acuerdo con la clasificación de la Organización Mundial de la Salud (OMS).1, 9 En el parénquima no tumoral, se evaluó la presencia de cirrosis, displasia hepatocítica, esteatosis e invasión vascular.
Las técnicas inmunohistoquímicas se realizaron sobre secciones histológicas de 3 micras mediante un sistema automatizado de inmunomarcación de acuerdo con las pautas del fabricante (Benchmark XT, Ventana). Se utilizaron los siguientes anticuerpos: CK7 (OV-TL 12/30; Cell Marque; monoclonal de ratón; 1/100), CK8/18 (B22.1-23.1; Cell Marque; monoclonal de ratón; 1/100), CK19 (RCK 108; bioGENEX; monoclonal de ratón; 1/100), CEA (CEA31; Cell Marque; monoclonal de ratón; 1/100), CD34 (QBEnd/10; Cell Marque; monoclonal de ratón; 1/100), E-cadherina (EP700Y; Cell Marque; monoclonal de conejo; 1/50), ß-catenina (Clone 14; Cell Marque; monoclonal de ratón; 1/50), Hepar-1 (OCH1E5, DAKO; monoclonal de ratón; 1/50), p53 (DO7; Cell Marque; monoclonal de ratón; 1/100), Glypican-3 (1G12; Cell Marque; monoclonal de ratón; 1/50) y Ki67 (SP6; Cell Marque; monoclonal de conejo; 1/100).
Los cortes histológicos fueron desparafinados y rehidratados. La recuperación antigénica se llevó a cabo mediante pretratamiento con altas temperaturas (60 minutos a 65°C). Para la detección y visualización se utilizó el kit ultraview universal DAB (ventana). La sustitución de todos los anticuerpos primarios por PBS fue utilizado como control negativo.
Para el análisis semicuantitativo de los marcadores de diferenciación epitelial (Hepar-1, CK8/18, CEA, CK7, CK19 y Glypican-3) se consideró como positivos a los tumores con más del 5% de expresión en células tumorales. Para los marcadores relacionados a la carcinogénesis (E-cadherina, ß-catenina y p53) se consideró como positivo focal (pérdida parcial) a la marcación en menos del 20% de la superficie tumoral y positiva difusa a aquella con expresión en más del 20% del tumor.
Se definió a los CHC como neoplasias de baja o alta fracción de proliferación tomando como punto de corte un Ki67 del 20%.
Análisis estadísticoLos resultados fueron presentados como media y error estándar o como porcentaje; se consideró como estadísticamente significativo una p < 0,05. Los resultados fueron analizados utilizando el test de Mann Whitney y/o ANOVA. Los gráficos y el análisis de los datos fueron realizados utilizando el Prism software GraphPad-6 (La Jolla, CA) y el MedCalc Statistical Software versión 14 (MedCalc Software bvba, Ostend, Belgium; http://www.medcalc.org).
Resultados
De las 126 piezas quirúrgicas hepáticas analizadas, 51 presentaban CHC, 38 de las cuales pertenecían a pacientes de género masculino (75%) y 13 (25%) al femenino. La edad media al momento de la cirugía fue de 63 (rango: 30-85 años).
Las características demográficas de los pacientes así como el agente etiológico asociado se muestran en la Tabla 1. La infección con el VHC se encontró en 20 pacientes (39%), seguida por la intoxicación crónica por alcohol (n = 7; 14%). Las causas menos frecuentes fueron VHB y HH en 2 pacientes respectivamente (4%), un caso de cirrosis biliar primaria y uno de hepatitis autoinmune (2% respectivamente). Cinco pacientes presentaron causas asociadas de CHC, 4 de ellos (8%) alcohol y VHC, y uno (2%) alcohol y HH. En trece casos (26%) la causa de CHC fue indeterminada (criptogénica).
Tabla 1. Datos clínicos y tipo de cirugía practicada en pacientes con CHC.

Estudio macroscópico de los CHCLos hallazgos de este estudio mostraron que el 70,6% de los casos (n = 36) presentaron una neoplasia única, mientras que en el resto (29,4%) los CHC fueron multifocales (2 a 3 tumores por espécimen). Se observó mayor tamaño en los tumores únicos con una media de 3,5 ± 0,5 cm de diámetro mayor (rango: 0,6-16,5), en comparación con los multifocales en donde se identificaron CHC de menor tamaño (rango: 0,5-12). La localización más frecuente de los CHC fue en el lóbulo hepático derecho (n = 34; 67%).
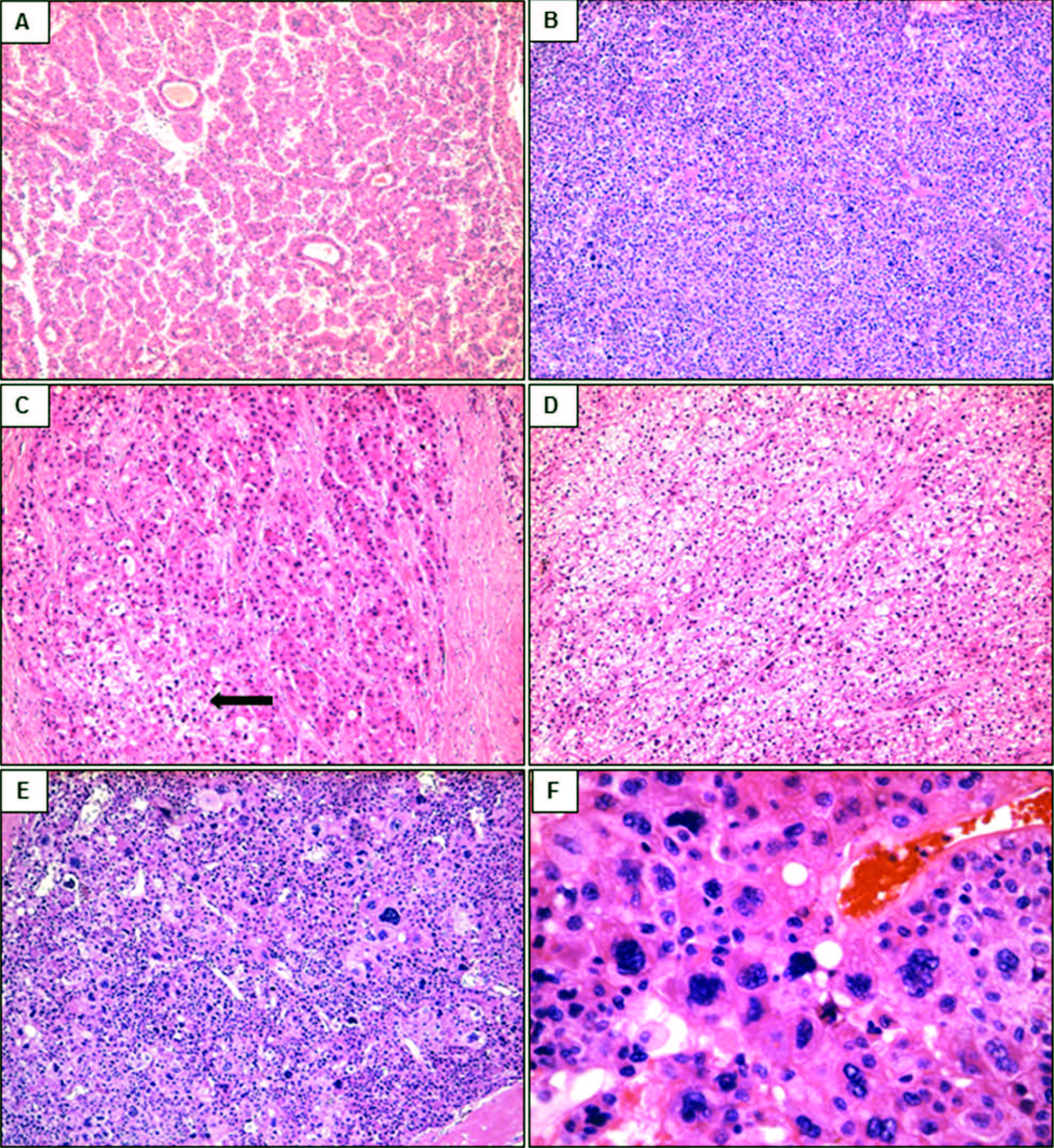
Estudio histológico de los CHC y del parénquima no tumoralEn el estudio con microscopía de luz, 22 neoplasias evidenciaron arquitectura trabecular (43%). Diecisiete tumores (33%) mostraron un patrón combinado trabecular y pseudoglandular u otras asociaciones (trabecular y sólido 10%; trabecular y células claras 4%) (Figuras 1A a 1D). Se encontraron cuatro CHC que combinaban hasta 3 patrones diferentes dentro del mismo tumor (trabecular, pseudoglandular y sólido 6%; trabecular, pseudoglandular y células claras 2%).
El grado histológico mayormente observado (n = 33; 65%) fue el moderadamente diferenciado (grado II), seguido por el bien diferenciado (grado I) (n = 13; 25%). Tres casos (6%) presentaron un grado histológico mixto, alternando zonas de CHC de grado II con grado III, con presencia de células gigantes pleomórficas (Figuras 1E y 1F). En nuestra serie el grado III se encontró siempre asociado con áreas de grado II y nunca como constituyente único del tumor. Se identificó invasión de la microvasculatura en el 41% (n = 21).
En el parénquima hepático no tumoral se observó que el 94% (n = 48) presentaba cirrosis asociada. El resto evidenció esteatosis macrovacuolar en 2 casos y un cuadro hepatítico con severa actividad.
Inmunotipificación de los CHCPara definir el fenotipo de las células neoplásicas, luego del estudio morfológico se realizaron técnicas inmunohistoquímicas, observándose positividad con CK8/18 en el 86% (n = 44) de los CHC, con Hepar-1 en el 69% (n = 35), con Glypican-3 en el 66,6% (n = 24) y con CEA en el 22% (n = 11). El CD34 remarcó el patrón sinusoido-trabecular de los CHC en el 100% de los tumores.
Por otra parte, se evidenció que la mayoría de los CHC conservaban la expresión de E-cadherina. Treinta y siete tumores (73%) mostraron positividad difusa y 8 (16%) evidenciaron positivad focal (pérdida parcial) de dicho marcador.
La ß-catenina mostró positividad nuclear en 10 tumores (20%), 8 (16%) de ellos con patrón de marcación difuso y 2 (4%) con positividad focal. La sobreexpresión de la proteína p53 se constató en el 24% de los CHC (n = 12), en todos los casos con una marcación difusa.
La fracción de proliferación tumoral demostró un rango muy amplio del 1% al 90% (media 17,6%), por lo que se tomó como punto de corte un Ki67 de 20%, observándose que el 61% de los CHC (n = 31) correspondían a neoplasias con baja fracción de proliferación, mientras que en el 27% (n = 14) la fracción de proliferación fue alta.
Figura 1. Patrones arquitecturales y citológicos de los CHC coloreados con hematoxilina y eosina. A: CHC con patrón de crecimiento trabecular y pseudoglandular. B: CHC de tipo sólido. C y D: CHC con patrón trabecular y sectores de células claras (flecha). E y F: CHC con patrón de crecimiento sólido y de células bizarras multinucleadas. Aumento original: 100X (A, B, C, D, E); 400X (F). CHC: carcinoma hepatocelular.

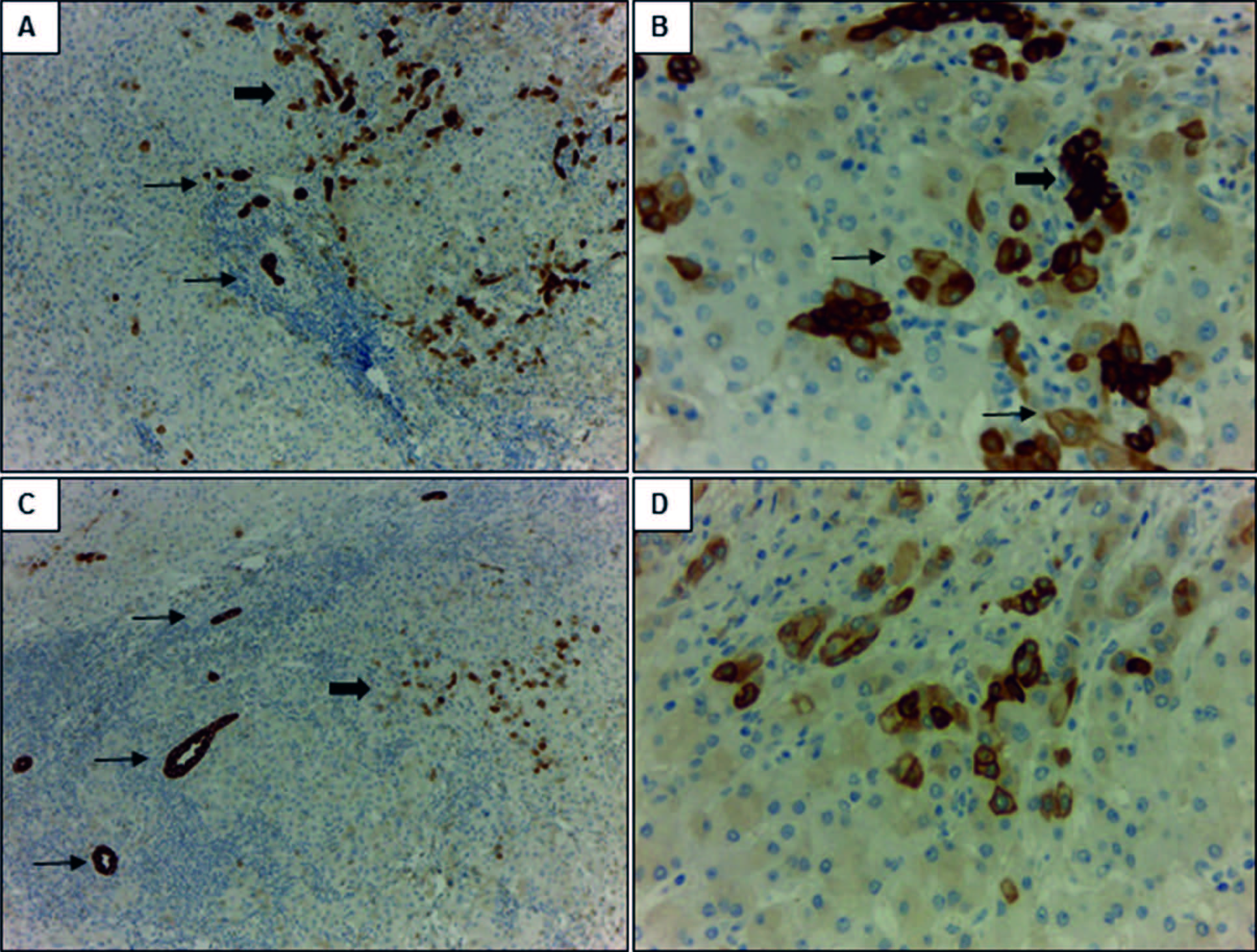
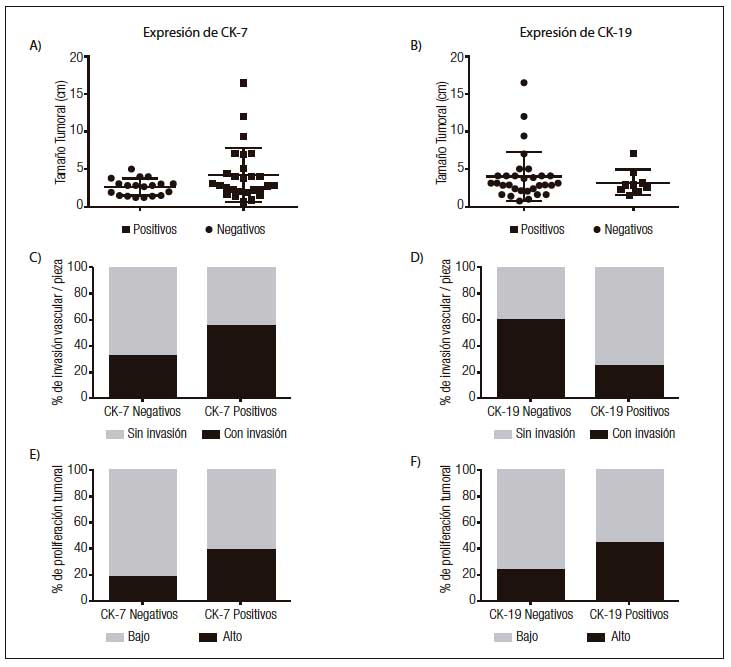
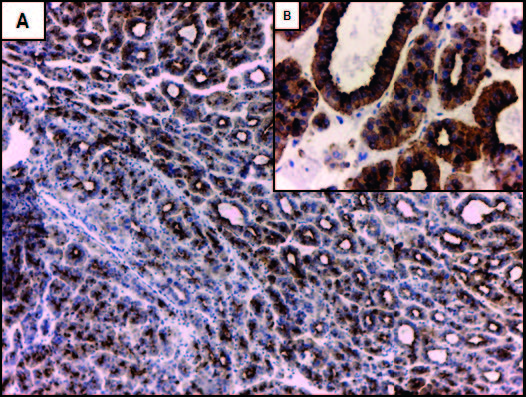
Expresión de CK7, CK 19 y Glypican-3 en CHCLos resultados del análisis por inmunohistoquímica mostraron que el 52,9% (n = 27) de los CHC expresaban CK7 (Figuras 2A y 2B). No se observó diferencia significativa entre los CHC CK7 positivos y negativos, en relación con el tamaño tumoral (12 de 27 CHC CK7 positivos y 5 de 19 CK7 negativos eran de gran tamaño, p = 0,16), la presencia de invasión vasculolinfática (15 de 27 CHC CK7 positivos y 6 de 19 CK7 negativos presentaban invasión vasculolinfática, p = 0,68) y la multifocalidad tumoral (8 de 27 CHC CK7 positivos y 6 de 19 CK7 negativos eran multifocales, p = 0,08) siendo en su mayoría unifocales (70,5%, n = 36). Sin embargo, en los casos CK7 negativos se evidenció una tendencia a una mayor proporción de CHC pequeños (< 3 cm) (14 tumores pequeños vs. 5 grandes) y a la ausencia de compromiso vasculolinfático (13 CHC sin vs. 6 con invasión vasculolinfática) en comparación a los CHC con positividad para CK7 (Figuras 3A y 3C). Más aún, los tumores de gran tamaño iguales o mayores a 7 cm (n = 6) fueron en todos los casos positivos para CK7.
Al comparar el índice de proliferación tumoral (Ki67) entre los CHC que expresaban o no CK7, se observó que los tumores positivos para dicho marcador presentaban una fracción proliferativa más alta a pesar de que la diferencia entre ambos grupos no fue significativa (9 de 27 CHC CK7 positivos y 4 de 19 CK7 negativos tenían alta proliferación, p = 0,17) (Figura 3E).
Figura 2. Expresión inmunohistoquímica de marcadores de linaje biliar en un carcinoma hepatocelular (CHC). A: la marcación con CK7 muestra un patrón de positividad en parches y con diferente intensidad en el CHC (flecha gruesa), en comparación con los conductos biliares reactivos peritumorales con marcación fuerte y homogénea (flecha fina). B: mayor aumento de A; positividad con CK7 en hepatocitos neoplásicos de tamaño pequeño que remedan a las células hepáticas progenitoras (CHP) (flecha gruesa) y en otros más grandes con mayor diferenciación (flecha fina). C: expresión focal de CK19 (flecha gruesa) en un CHC con marcada proliferación ductular peritumoral (flecha fina). La marcación en los conductos biliares proliferados es más intensa en comparación con los hepatocitos neoplásicos. D: mayor aumento de C; marcación de membrana en hepatocitos del CHC. Aumento original: 100X (A, C); 400X (B, D).

Al analizar la marcación con CK19, se observó que el 17,6% (n = 9) expresaban dicho marcador (Figuras 2C y 2D). No se evidenció diferencia significativa en los CHC positivos y negativos para CK19 en relación con su tamaño tumoral (2 de 9 CHC CK19 positivos y 14 de 31 CK19 negativos eran de gran tamaño, p = 0,51), la invasión vascular (2 de 9 CHC CK19 positivos y 18 de 31 CK19 negativos presentaban invasión vasculolinfática, p = 0,66), la multifocalidad (3 de 9 CHC CK19 positivos y 11 de 31 CK19 negativos eran multifocales, p = 0,96) (Figuras 3B y 3D). Si bien en ambos grupos predominaron los CHC con bajo índice de proliferación, se observó una tendencia al aumento de la fracción proliferativa en aquellos tumores que expresaban CK19 (4 de 9 CHC CK19 positivos y 8 de 31 CK19 negativos tenían alta proliferación, p = 0,22) (Figura 3F).
Por otra parte, se observó que el 66,6% de los CHC expresaban Glypican-3 (n = 24) (Figura 4). No se identificó diferencia significativa entre los grupos positivos y negativos para CK7 y CK19 en relación a la expresión de Glypican-3 (p = 0,15 y p = 0,6, respectivamente).
Tampoco se demostró diferencia significativa en la sobrevida de los pacientes al analizar dichos grupos (18 y 8 pacientes con CHC CK7 y 19 positivos, así como 15 y 20 pacientes con CHC CK7 y 19 negativos, respectivamente, están vivos en la actualidad).
Figura 3. Tamaño tumoral (A y B), porcentaje de invasión vascular (C y D) e índice de proliferación (E y F) en carcinomas hepatocelulares, en relación con la expresión de CK7 y CK19.

Figura 4. A: expresión inmunohistoquímica de Glypican-3 en un carcinoma hepatocelular (CHC) con patrón pseudoglandular y marcación citoplasmática. B: mayor aumento de (A). Aumento original: 100X (A); 400X (B).

Discusión
En el presente trabajo se estudió una serie argentina de CHC desarrollados en hígados con diversas patologías asociadas. Si bien dichas neoplasias han sido bien caracterizadas en ciertos países, poco se sabe de los CHC en nuestra región.2, 5, 6 Los datos epidemiológicos recopilados en nuestra serie fueron similares a los descriptos en un trabajo argentino realizado por Fassio y col. Mayor frecuencia de CHC en el género masculino, edad media cercana a los 63 años y acompañados de cirrosis en más del 90% de los casos. Sin embargo, en lo referente a la etiología, en nuestra serie el VHC llegó casi a triplicar el alcoholismo crónico (39% y 14% respectivamente), contrariamente al trabajo previamente citado, en donde la intoxicación crónica por alcohol predominó ligeramente en relación con el VHC (41,6% y 40,5% respectivamente).5 Según las distintas publicaciones, las proporciones de VHC varían en relación con la distribución geográfica, asemejándose la nuestra a la encontrada en Italia (44%- 66%), Francia (27%-58%) y a la reportada en un estudio latinoamericano que incluyó a la Argentina (30,8%).2, 6
En el presente trabajo, reportamos por primera vez en Argentina datos que corroboran la heterogeneidad morfológica de estos tumores, evidenciando un alto porcentaje de neoplasias combinadas, con diferentes patrones histológicos y grado de diferenciación dentro del mismo tumor. Trabajos recientes remarcaron la importancia de complementar el estudio morfológico de los CHC con el análisis inmunofenotípico, con la intención de responder a distintos interrogantes tales como la célula de origen tumoral, su comportamiento y, en un futuro, su sensibilidad al tratamiento.21
Dado que los CHC derivan de hepatocitos transformados, es esperable que estas células expresen marcadores de dicho linaje. Sin embargo, en algunos CHC ha sido reportada la coexpresión de marcadores hepatocíticos y biliares (CK7 y CK19), como resultado de la desdiferenciación de los hepatocitos hacia estadios más inmaduros con potencial bifenotípico.10, 22, 23 Estos hallazgos llevaron a proponer que algunos CHC se originarían en las CHP o stem cells, cuya histogénesis es aún desconocida, pero que se asociarían a un peor pronóstico.10 A pesar de la limitación en el tamaño muestral, consideramos que los resultados del inmunoperfil aquí analizado son de gran interés. Por un lado, se observó que casi el 53% de los CHC expresaban CK7, llegando casi a triplicar la marcación con CK19. Esta marcación resultó, además, mayor a la observada en otras series, lo que corrobora la heterogeneidad de dichos tumores no solo a nivel morfológico, sino también en su inmunofenotipo. Si bien en nuestra serie, al igual que en otros reportes, el tamaño y el compromiso vascular no tuvieron correlación con la expresión de CK7, se pudo evidenciar una tendencia hacia un comportamiento menos agresivo en los CHC CK7 negativos, dado que en su mayoría fueron más pequeños y carecían de invasión vascular.10, 22De manera similar, se pudo observar que, si bien la diferencia no fue significativa, la mayoría de los CHC CK7 positivos expresaban Glypican-3, un marcador utilizado en la actualidad para diferenciar lesiones hepatocíticas benignas de malignas.
Por otro lado, el mayor tamaño y el tipo de muestra (resecciones quirúrgicas parciales/biopsias hepáticas) analizado en otros reportes podría explicar las diferencias con los CHC aquí estudiados, provenientes en su mayoría de pacientes trasplantados y con neoplasias de menor tamaño. Más aun, en nuestra casuística, los únicos casos resecados parcialmente por su gran tamaño mostraron marcación positiva para CK7, con lo que se podría hipotetizar que en los CHC avanzados existiría una tendencia al aumento de la expresión de CK7.
Asimismo, pudimos evidenciar una tendencia al aumento del Ki67 en los CHC que expresaban CK7 y/o CK19. Estos resultados estarían de acuerdo con publicaciones previas que proponen que este subtipo de CHC con expresión de marcadores biliares tendría peor pronóstico.10, 22
Finalmente, incluimos en este estudio, el análisis inmunohistoquímico de ciertas proteínas implicadas en la vía de la carcinogénesis hepatocítica, en un intento de explicar los resultados controversiales previamente reportados.19, 20, 24, 25 Debido a la pérdida de E-cadherina en los distintos tumores epiteliales, se creyó que dicha molécula podría estar disminuida en los tumores hepatocíticos y de esta manera explicar su comportamiento agresivo.14-16 Sin embargo, en los CHC los resultados fueron divergentes, con pérdida de su expresión en algunos reportes y conservación en otros, incluida esta serie.17, 26 Hallazgos igualmente controversiales se encontraron al estudiar la alteración de la ß-catenina en los CHC, con un rango muy amplio de mutación según las distintas series (13% al 43%) e incluso en tumores hepáticos benignos.24-30
El análisis de la sobreexpresión de la proteína p53 tendría un interés epidemiológico suplementario, debido a su relación con distintos agentes etiológicos relacionados al CHC, como la exposición a la aflatoxina B1 y su asociación con la HH.19, 20 En nuestro estudio, la sobreexpresión de la proteína p53 se constató en el 24% de los CHC, 2 de los cuales presentaban HH como factor asociado. Nuevos estudios con mayor número de casos serían necesarios para esclarecer la expresión de estas 3 moléculas en los CHC de nuestra región.
Conclusión
Describimos por primera vez en nuestra población la morfología y el inmunoperfil de una serie de CHC. Evidenciamos un alto porcentaje de CHC con positividad para CK7 y CK19 con tendencia al aumento de su fracción proliferativa. Los CHC negativos para CK7 fueron mayormente de pequeño tamaño y no mostraron invasión vascular, por lo que podrían señalar un subgrupo de CHC provenientes de una CHP con potencial bifenotípico y comportamiento biológico diferente. Los CHC positivos para CK7 mostraron una tendencia a la mayor expresión de Glypican-3, marcador relacionado con la malignidad hepatocítica.
La relevancia e incidencia de la expresión de CK7 y CK19 en CHC no ha sido aún explorada en series latinoamericanas. Estos resultados, en conjunto con los hallazgos controversiales en relación con la expresión de E-cadherina, ß-catenina y p53, requieren estudios colaborativos y con un mayor tamaño muestral, con el fin de dilucidar factores de mal pronóstico o predictores de recurrencia.
Dado que en nuestra serie la mayoría de los CHC se asociaban al VHC, sería de gran interés ampliar el estudio a los casos asociados con otros factores etiológicos, con el objetivo de promover la vigilancia de los pacientes en riesgo y de este modo detectar precozmente la aparición de estos tumores.
Sostén financiero. El soporte financiero para la publicación del presente manuscrito proviene del Premio de Honor al Trabajo Científico de Investigación 2014, otorgado por la Asociación de Médicos y Odontólogos del Hospital Británico de Buenos Aires.
Agradecimientos. Los autores agradecen a Olga Ocampo por el soporte técnico y a Mara Canale y Yanina Sivrigian por la colaboración administrativa.
Conflicto de intereses. Los autores declaran que no existen conflictos de interés.



{kind=link}
{kind=link}